As well as in depth metabolomic analysis, SMEW also supports the integration of multimodal data, such as histology images (as detailed in the histology vignette) and bulk-level omics data such as transcriptomics or proteomics. This allows users to explore spatial metabolomics data in the context of other molecular or imaging modalities, providing richer biological insights and enabling more comprehensive analyses.
SMEW’s multi-omic integration is performed through multi-modal networks calculating covariation between metabolites, genes, and/or proteins at the sample level.
Input data format for multi-omic integration
To perform multi-omic integration in SMEW, you will need to provide an additional input file for your bulk-level omics data (e.g. transcriptomics or proteomics).
The data should be saved as a csv file with features (e.g. genes, proteins etc.) on rows and samples (which must match entries in the main object metadata Sample column) on columns like follows:
| id | sample_1 | sample_2 | sample_3 |
|---|---|---|---|
| gene_1 | 23.4 | 13.3 | 77.9 |
| gene_2 | 60.7 | 56.9 | 43.1 |
| gene_3 | 113.4 | 100.8 | 112.2 |
| gene_4 | 12.5 | 10.8 | 46.7 |
| gene_5 | 89.9 | 50.3 | 79.3 |
| gene_6 | 54.4 | 30.9 | 46.0 |
| … | … | … |
Performing multi-omic integration in SMEW
To perform multi-omic integration in SMEW, simply include your
bulk-level omics data file when creating your app with
create_smew_app().
In the ‘Multi-modal covariation network inference’ tab, you can infer covariation networks between features in the MSI data and features in the bulk-level data. This allows users to explore relationships between different types of data and identify multi-modal patterns of variation that may be relevant to their biological question. Here the peak node colours remain the same as the previous network, while other modality targets are shown in light pink, overlapping other modality peaks are shown in dark pink and non-overlapping other modality peaks are shown in purple.
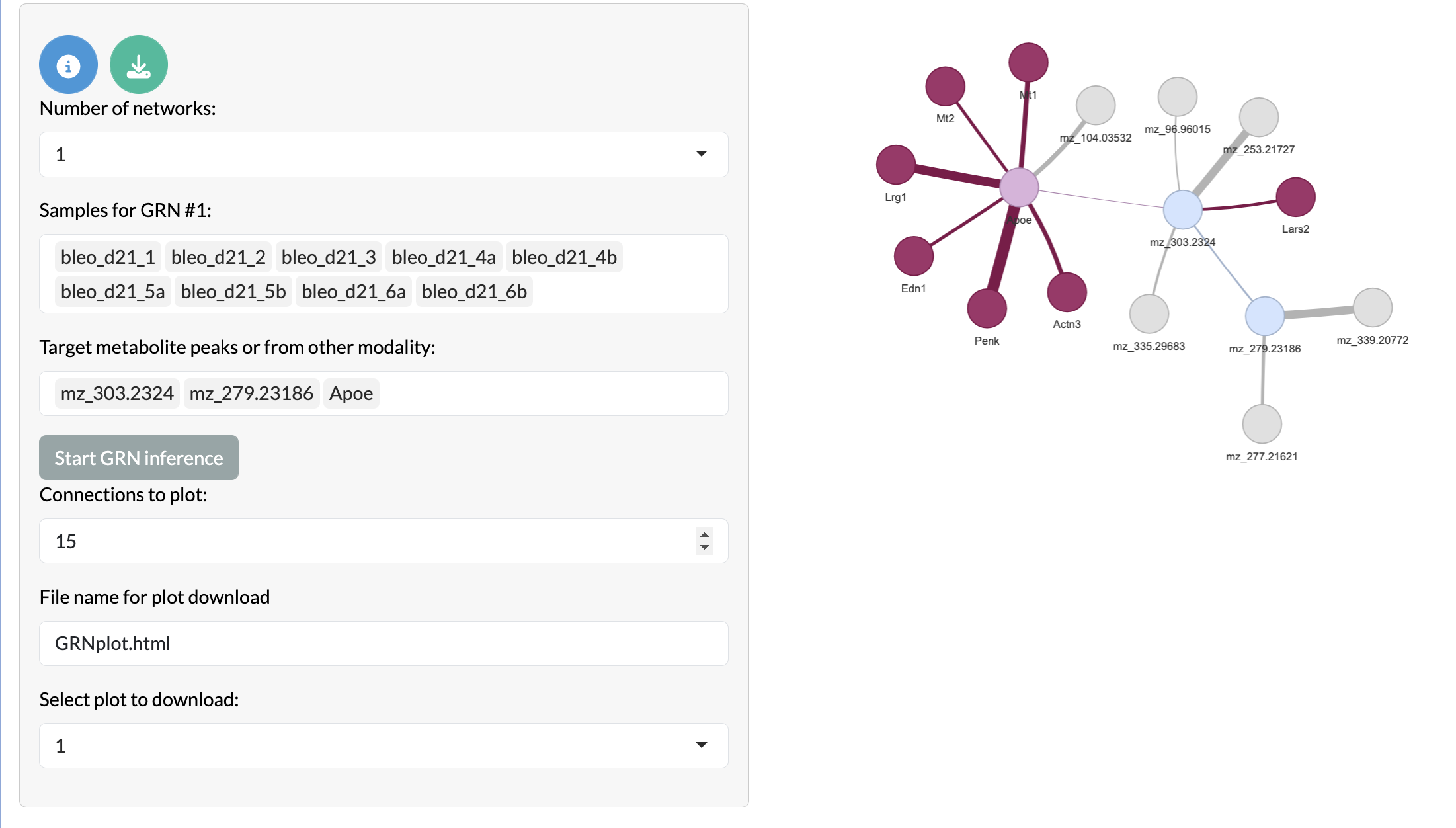
Multi-modal covariation network inference
It is worth bearing in mind that covariation networks can be dominated by the larger modality and so it may be a good idea to limit the number of features in the bulk-level data to the most variable or most relevant features to ensure that the resulting network is not dominated by the comparatively large number of genes or proteins compared to metabolite peaks.