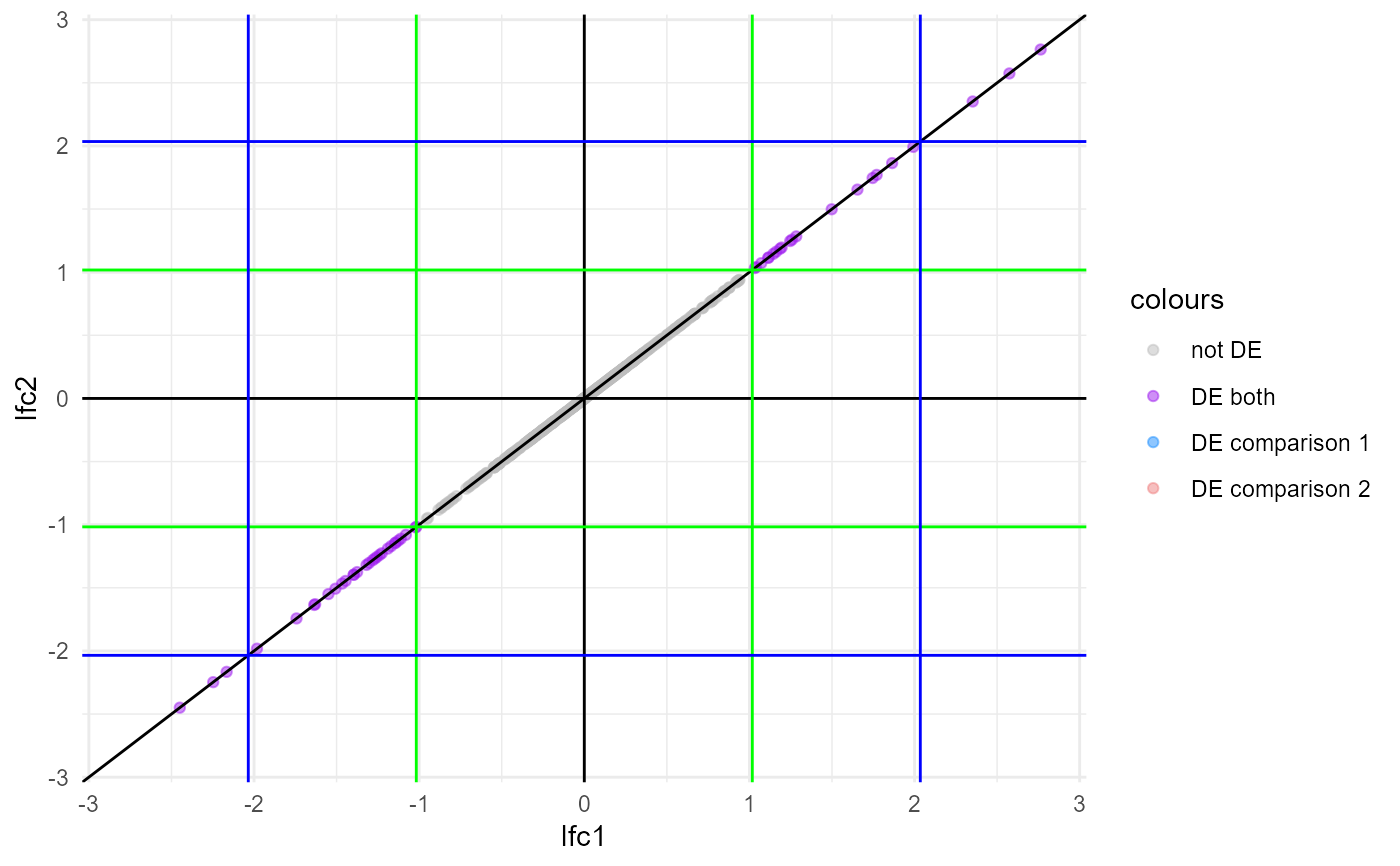
Create a cross plot comparing differential expression (DE) results
Source:R/cross_plot.R
cross_plot.RdThis function creates a cross plot visualising the differences in log2(fold-change) between two DE analyses.
Usage
cross_plot(
DEtable1,
DEtable2,
DEtable1Subset,
DEtable2Subset,
df = NULL,
lfc.threshold = NULL,
raster = FALSE,
mask = FALSE,
labnames = c("not DE", "DE both", "DE comparison 1", "DE comparison 2"),
cols.chosen = c("grey", "purple", "dodgerblue", "lightcoral"),
labels.per.region = 5,
fix.axis.ratio = TRUE,
add.guide.lines = TRUE,
add.labels.custom = FALSE,
genes.to.label = NULL,
seed = 0,
label.force = 1
)Arguments
- DEtable1, DEtable2, DEtable1Subset, DEtable2Subset
tables of DE results, usually generated by
DEanalysis_edger; the first two should contain all genes, while the second two should only contain DE genes- df
Optionally, pre-computed cross plot table, from cross_plot_prep
- raster
whether to rasterize non-DE genes with ggraster to reduce memory usage; particularly useful when saving plots to files
- mask
whether to hide genes that were not called DE in either comparison; default is FALSE
- labnames, cols.chosen
the legend labels and colours for the 4 categories of genes ("not DE", "DE both", "DE comparison 1", "DE comparison 2")
- labels.per.region
how many labels to show in each region of the plot; the plot is split in 8 regions using the axes and major diagonals, and the points closest to the origin in each region are labelled; default is 5, set to 0 for no labels
- fix.axis.ratio
whether to ensure the x and y axes have the same units, resulting in a square plot; default is TRUE
- add.guide.lines
whether to add vertical and horizontal guide lines to the plot to highlight the thresholds; default is TRUE
- add.labels.custom
whether to add labels to user-specified genes; the parameter genes.to.label must also be specified; default is FALSE
- genes.to.label
a vector of gene names to be labelled in the plot; if names are present those are shown as the labels (but the values are the ones matched - this is to allow custom gene names to be presented)
- seed
the random seed to be used for reproducibility; only used for ggrepel::geom_label_repel if labels are present
- label.force
passed to the force argument of ggrepel::geom_label_repel; higher values make labels overlap less (at the cost of them being further away from the points they are labelling)
- pval.threshold
the log2(fold-change) threshold to determine whether a gene is DE
Examples
expression.matrix.preproc <- as.matrix(read.csv(
system.file("extdata", "expression_matrix_preprocessed.csv", package = "bulkAnalyseR"),
row.names = 1
))[1:500, 1:4]
anno <- AnnotationDbi::select(
getExportedValue('org.Mm.eg.db', 'org.Mm.eg.db'),
keys = rownames(expression.matrix.preproc),
keytype = 'ENSEMBL',
columns = 'SYMBOL'
) %>%
dplyr::distinct(ENSEMBL, .keep_all = TRUE) %>%
dplyr::mutate(NAME = ifelse(is.na(SYMBOL), ENSEMBL, SYMBOL))
#>
#> 'select()' returned 1:many mapping between keys and columns
edger <- DEanalysis_edger(
expression.matrix = expression.matrix.preproc,
condition = rep(c("0h", "12h"), each = 2),
var1 = "0h",
var2 = "12h",
anno = anno
)
deseq <- DEanalysis_edger(
expression.matrix = expression.matrix.preproc,
condition = rep(c("0h", "12h"), each = 2),
var1 = "0h",
var2 = "12h",
anno = anno
)
cross_plot(
DEtable1 = edger,
DEtable2 = deseq,
DEtable1Subset = dplyr::filter(edger, abs(log2FC) > 1, pvalAdj < 0.05),
DEtable2Subset = dplyr::filter(deseq, abs(log2FC) > 1, pvalAdj < 0.05),
labels.per.region = 0
)